pseudoBulk
Cluster-specific pseudo-bulk analysis of 10X single-cell RNA-seq data by connecting Seurat to the VBC RNA-seq pipeline.
This project is maintained by vertesy
Cluster-specific pseudo-bulk analysis of 10X single-cell RNA-seq data
Cluster-specific pseudo-bulk analysis of 10X single-cell RNA-seq data by connecting to the VBC RNA-seq pipeline.
Introduction
See Document on pseudo-bulk analysis and my presentation on pseudo-bulk analysis. In brief, pseudo-bulk analysis allows
- top-level overview & comparison of all single-cell datasets generated in the lab
- comparison single-cell-clusters vs bulk (sorted) RNA-seq samples
- in depth study of sequencing libraries (read position distributions, extensive quality control).
- Separation of .bam files per sample (in case of sample pooling, e.g. MULTI-SEQ)
Pseudo-bulk analysis for top-level overview
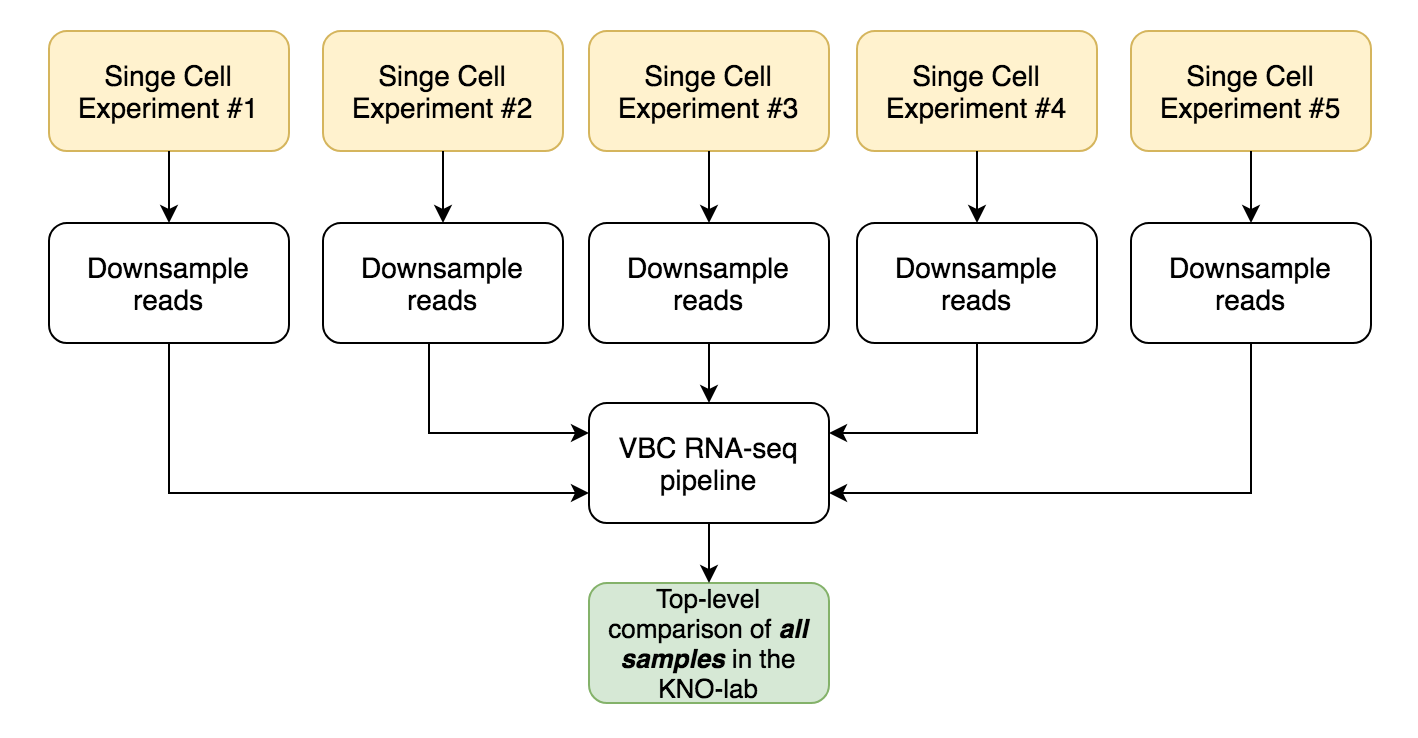
Top-level overview is very simple and does not require preprocessing in R.
Pseudo-bulk analysis for cluster-specific comparisons
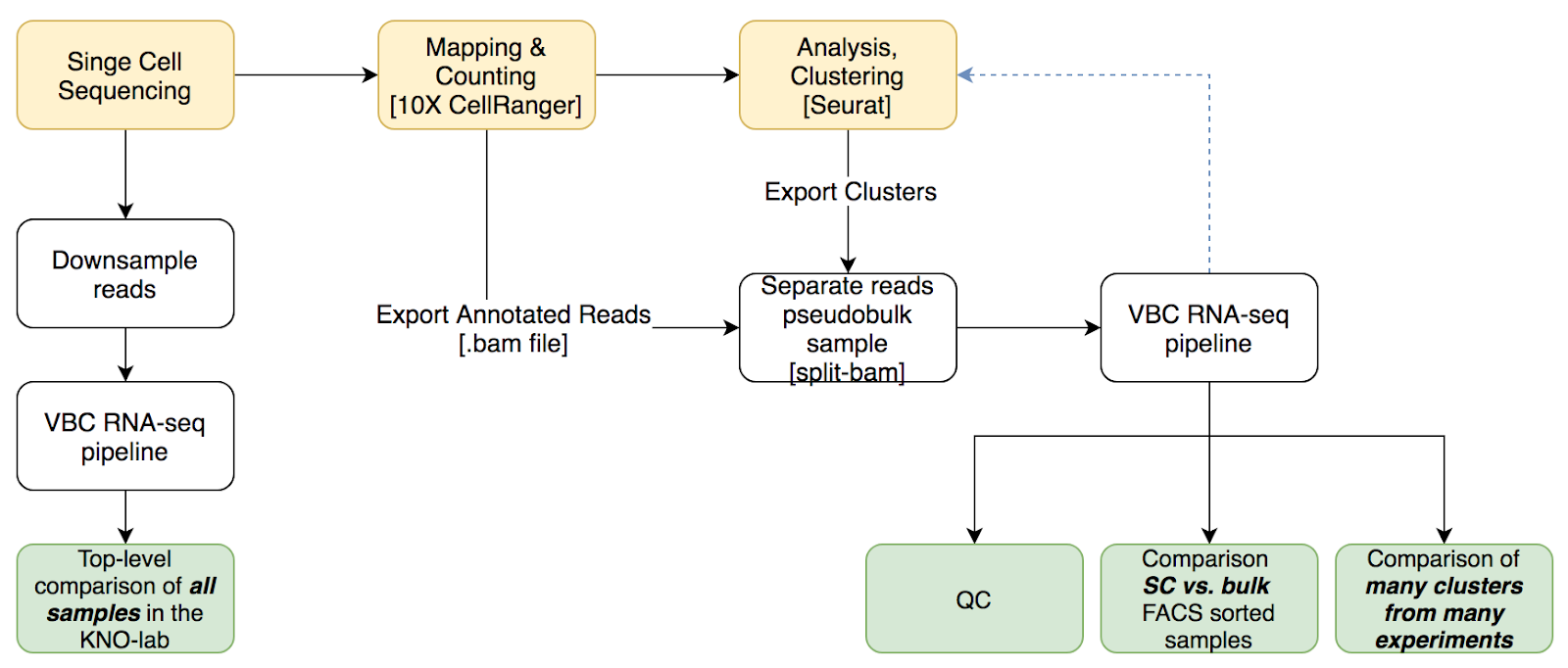
Cluster-specific overview requires preprocessing in R and the export of barcodes. The exported barcodes then will be processed by
Steps for the cluster-specific analysis
1. Run SeuratFind clusters, etc.
[explained elsewhere]
##2 Export clusters
Depends: CodeAndRoll.R, and MarkdownReports
Use get.ls.of.CBCs and write.out.CBCs.per.cl to write out the barcodes per dataset, per cluster. Script found in: /Users/abel.vertesy/GitHub/Seurat.multicore/Seurat3.Write.Out.CBCs.for.subset-bam.R
get.ls.of.CBCs <- **function**(scobj = combined.obj, ident = 'integrated_snn_res.0.3',
plotit=T, trim.libName=F) { #
Idents(scobj) <- ident
id_x = Idents(scobj)
dsets = unique(stringr::str_split_fixed(names(id_x), pattern = '_', n = 2)[,2])
iprint("Returns a list of lists (",l(dsets),"libraries [",dsets,"] /",
l(levels(id_x)),"clusters [",levels(id_x),"] )")
ls_CBCs = list.fromNames(levels(id_x))
print("Cluster:")
**for** (cl **in** 1:l(levels(id_x))) {
print(cl)
cells_x = WhichCells(combined.obj, idents = id_x[cl])
cells_perLib = stringr::str_split_fixed(cells_x, pattern = '_', n = 2)
cells_perLib = cbind(cells_perLib,scobj$orig.ident[cells_x])
ls_CBCs[[cl]] <- ls_cells_clX_perLib <- split(x = cells_perLib, f = cells_perLib[,3])
}
revlist = reverse.list.hierarchy(ls_CBCs)
**if** (!isFALSE(trim.libName)) { # remove .WT **and** .TSC2
names(revlist) = stringr::str_split_fixed(names(revlist), pattern = "\\.", n=2)[,1]
}
**if** (plotit) {
**for** (i **in** 1:l(revlist)) {
ClusterSizes = unlapply(revlist[[i]], l)
wpie(ClusterSizes, savefile = F, plotname = paste('ClusterSizes, cl.', i))
}
}
**return**(revlist)
}
# ls.of.CBCs = get.ls.of.CBCs(trim.libName = T); names(ls.of.CBCs)
# *------------------------------------------------------------*
write.out.CBCs.per.cl <- **function**(ls_CBCs = ls.of.CBCs, add.suffix="-1", openOutDir=T) { # take the output of get.ls.of.CBCs() as input, **and** write out as csv
(depth = l(ls_CBCs))
(dsets = names(ls_CBCs))
**for** (i **in** 1:l(dsets)) {
outputDir = p0(OutDir,"CBCs/", dsets[i])
dir.create(outputDir, recursive = T)
inside.ls = ls_CBCs[[i]]
**for** (j **in** 1:l(inside.ls)) {
CBCs = inside.ls[[j]]
**if** (!isFALSE(add.suffix)) CBCs = p0(CBCs,add.suffix)
write.simple.vec(input_vec = CBCs, ManualName = p0(outputDir,"/Cl.", j, ".csv"))
}
}
**if** (openOutDir) system(paste("open", outputDir))
}
# write.out.CBCs.per.cl()
3. Copy output folder next to bam folder
Example folder structure:

4 Subset bam by run.split-bam.sh
Barcoded BAM is described here. Alternative method is described here
Depends: 10XGenomics/subset-bam
Script found in: /Users/abel.vertesy/GitHub/Seurat.multicore/split.bam.files.scripts/run.split-bam.sh:
# https:*//github.com/10XGenomics/subset-bam*
# alternative scripts: https:*//divingintogeneticsandgenomics.rbind.io/post/split-a-10xscatac-bam-file-by-cluster/*
# --bam (-b): **Input** 10x Genomics BAM. This BAM must have the CB tag to define the barcodes of cell barcodes (or the tag defined **by** --bam-tag). Must also have **an** index (.bai) **file**. REQUIRED.
# --cell-barcodes (-c): A cell barcodes **file** **as** produced **by** Cell Ranger that defines **which** barcodes were called **as** cells. **One** barcode per **line**. **In** Cell Ranger runs, this can be found **in** the sub-folder **outs**/filtered_gene_bc_matrices_mex/${refGenome}/barcodes.tsv where ${refGenome} is the name of the reference genome used **in** your Cell Ranger **run**. This **file** can be used **as** column labels **for** the output **matrix**. REQUIRED.
# --**out**-bam (-o): A path to write the subsetted BAM **file** to. REQUIRED.
# --cores: Number of parallel cores to **use**. DEFAULT: 1.
# --**log**-level: **One** of info, **error** or debug. Increasing **levels** of logging. DEFAULT: **error**.
# --bam-tag: Change this to **use** **an** alternative tag to the default CB tag. This can be useful **for** subsetting BAMs from LongRanger.
"on server"
tmux
cdd /groups/knoblich/users/abel/Data/
# lib="101147"
lib="101146"
bamdir="bam.files.cellranger/"$lib
bamfile=$bamdir/$(**ls** $bamdir | grep ".bam$")
outdir=$bamdir"/bam.per.cl/"
**mkdir** $outdir
BCdir="CBCs/"$lib"/"
**for** BC **in** $(**ls** $BCdir | grep ".csv")
**do**
BCfile=$BCdir$BC
echo $BCfile
echo $bamfile
outbam=$outdir$BC".bam"
echo $outbam
subset-bam --cores 8 --bam $bamfile --cell-barcodes $BCfile --**out**-bam $outbam --**log**-level debug
done
# samtools **view** /Volumes/abel/Data/bam.files.cellranger/101146/bam.per.**cl**/**Cl**.1.csv.bam | head
# samtools **view** /Volumes/abel/Data/bam.files.cellranger/101146/Oli.d110.101146.WT.bam | head -100
##5 Reanalyzing with the VBC RNA-seq pipeline
Described elsewhere